
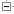
Gene/Protein Tree
Ensembl gene trees are generated by the Gene Orthology/Paralogy prediction method pipeline. All homologues in Ensembl are determined from gene trees.
Gene trees are constructed using one representative protein for every gene in every species in Ensembl. The longest translation annotated by the CCDS project is used, if any are available, or the longest protein-coding translation otherwise. (The trees can also be considered as protein trees).
The display shows the maximum likelihood phylogenetic tree representing the evolutionary history of genes. These trees are reconciled with a species tree, generated by TreeBeST. Internal nodes are then annotated for duplication (red boxes) or speciation (blue boxes) events.
Red squares represent duplications nodes, blue squares represent speciation nodes, giving rise to paralogues and orthologues. Another class of node, ambiguous, is shown as a lighter blue square.
The gene of interest is highlighted in red and within-species paralogues are shown in blue, if the option to view paralogues is selected (below the tree diagram).
Taxonomy IDs refer to the NCBI Taxonomy Browser. The number at the top of pop-up menus (upon clicking on a node) corresponds to the node_id from the protein_tree_node table in the compara database.
Multiple alignment of the peptides (green bars) was made using MUSCLE. Green bars shows areas of amino acid alignment, white areas are gaps in the alignment. Dark green bars indicate consensus alignments.
Click on a node to expand a collapsed set of branches into a full tree. The consensus amino acid alignment corresponds to the consensus residues in the collapsed node, and will be expanded when the tree is expanded.
You can also view a detailed sequence alignment in Wasabi by clicking on a node.
You can also use the collapse and expand links at the bottom as follows:
View current gene only: Shows the default view of the gene tree, where the selected gene and the node it is within is expanded fully, while all other nodes are collapsed.
View paralogs of current gene: Shows the current gene and all its paralogues with their nodes expanded fully, while all other nodes are collapsed.
View all duplication nodes: Expands all the red duplication nodes and all of the nodes they fall within, while speciation nodes remain collapsed.
View fully expanded tree: Expands all nodes.
Collapse all the nodes at the taxonomic rank: Choose your preferred taxonomic rank from the drop down to see all nodes expanded above that rank, and collapsed below it.
Configure this page to customise the tree. Colouring by clade can be removed.
Gene trees can be exported as EMF (Ensembl Multi Format) files from the Ensembl ftp site.